
Help:
How ADAM works:
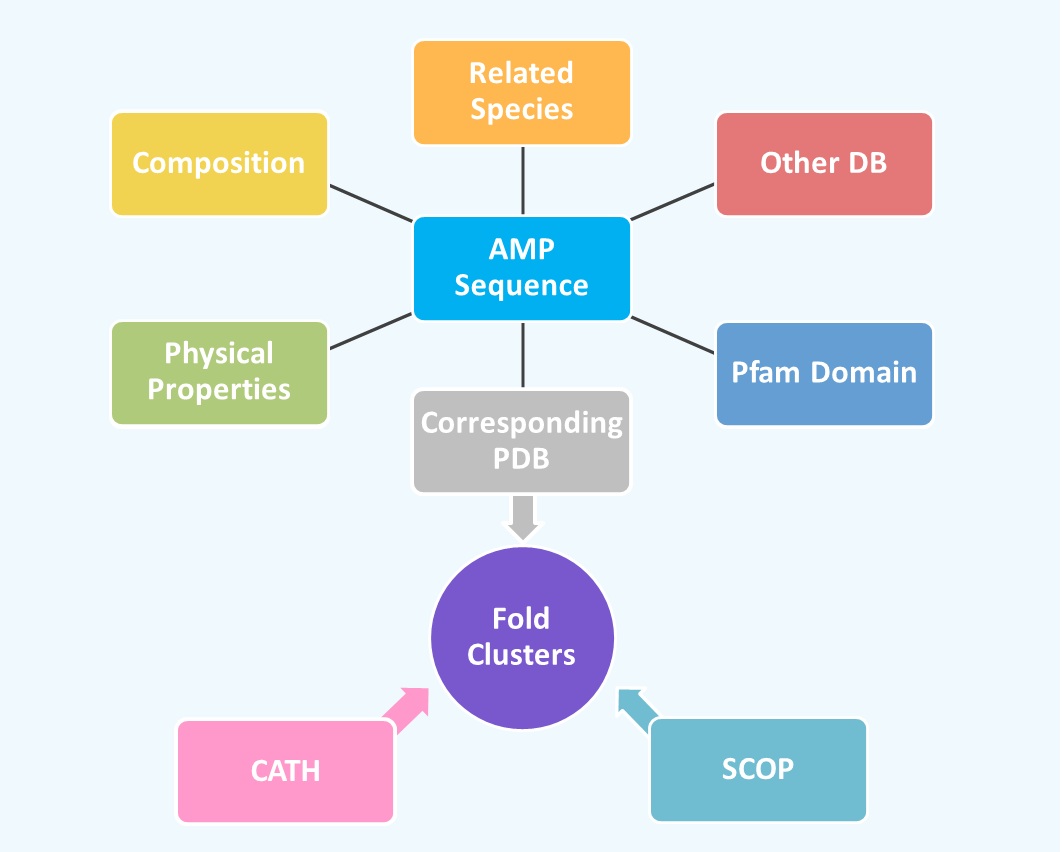
DB Statistics
Related Species
Average Composition Average Physical Properties Length Aliphatic Index Instability Index Net Charge Hydropathicity 39.56 89.95 34.30 +3.02 -0.06
Amino Acid Composition (AAC):Amino acid composition is the ratio of each amino acid in a peptide. The ratio of
an amino acid with type T in a peptide X is calculated as follows: 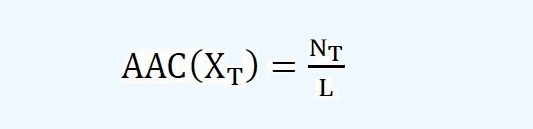
where NT is the number of the amino acid with type T and L is the length of peptide X. Aliphatic Index: The aliphatic index, the relative volume of aliphatic residues in a peptide, is
calculated as follows: 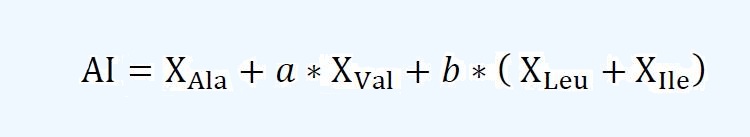
where a and b are the constants, which represent the relative volume of valine
and leucine or isoleucine to alanine. XAla, XVal, XLeu, and XIle are the fractions
of alanine, valine, leucine and isoleucine multiplied by 100, respectively. Instability Index: The instability index, an estimate of peptide stability, is calculated as follows: 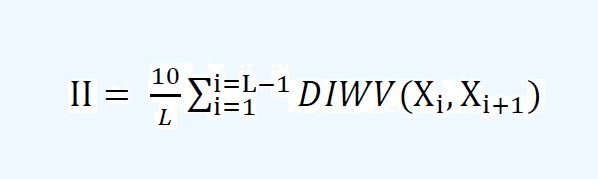
where L is the length of peptide and DIWV from the study by Guruprasad et al.
is an instability weight value of a dipeptide starting at position i . Peptides
with II values greater than 40 are considered to be unstable. Hydropathicity: Grand average of hydropathicity index (GRAVY) is used to represent the hydrophobicity
value of a peptide, which calculates the sum of the hydropathy values of all the amino acids divided by the sequence length. GRAVY was calculated using the hydropathy values
from Kyte and Doolittle. Search
ADAM offers multiple search capabilities, which can be classified into two basic
categories: sequence search and structural search. One can search what kind of
information an AMP sequence belongs to (sequence search) or which related
sequences are in a unique AMP structure or structural fold cluster (structural search).
Sequence search:
Search Type Description ADAM ID To search AMP sequences by ADAM ID Name To search AMP sequences by protein name Sequence To search AMP sequences by peptide segments Length To search AMP sequences by the chosen range of sequence length Taxonomy To search AMP sequences by selecting one of the seven species types Other DB To search AMP sequences which are also in other AMP database Pfam Domain To search ADAM sequences by Pfam domain name
Structural search:
Search Type Description Fold Cluster ID To search related AMP structural clusters by ADAM fold ID PDB ID To search AMP structural cluster by PDB ID
Browse clusters
To examine AMP sequence-structure relationships, ADAM allows users to browse through
the fold clusters built using TM-score. Each fold cluster gethers together the AMP
structures with the same structral fold defined by TM-score. Each cluster would
list the number of AMP sequences and the Pfam sequence associated with the fold.
A representative structure would be chosen by PDBselect to display its CATH and
SCOP annotation (if any). Any structures related to a particular fold defined by
CATH or SCOP can be displayed through their hyperlinks. More detail information for
the AMP sequences in the cluster can be displayed by clicking the cluster icon. Here
each structure in the cluster would be annotated by CATH, SCOP, and Pfam (if any). Prediction
In the ADAM database, we provide two different tools to examine whether the peptides or proteins are antimicrobial. Users can upload protein sequences with FASTA format to ADAM
to make prediction.
SVM:
Support vector machines are used to analyze AMP data and recognize patterns. Users are allowed to paste sequences in FASTA format to run our SVM models.
HMM:
HMMER is used to search any AMP homologs in the given protein sequence. It implements a probabilistic model called profile hidden Markov models (profile HMMs) to make predictions. Users are allowed to enter sequences in FASTA format to run our HMM models.
Links Other database links:
| How ADAM works: | |||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||
| DB Statistics | |||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||
Amino Acid Composition (AAC): | |||||||||||||||||||||||||||||||||||
| Amino acid composition is the ratio of each amino acid in a peptide. The ratio of an amino acid with type T in a peptide X is calculated as follows: | |||||||||||||||||||||||||||||||||||
where NT is the number of the amino acid with type T and L is the length of peptide X. | |||||||||||||||||||||||||||||||||||
| Aliphatic Index: | |||||||||||||||||||||||||||||||||||
| The aliphatic index, the relative volume of aliphatic residues in a peptide, is calculated as follows: | |||||||||||||||||||||||||||||||||||
where a and b are the constants, which represent the relative volume of valine and leucine or isoleucine to alanine. XAla, XVal, XLeu, and XIle are the fractions of alanine, valine, leucine and isoleucine multiplied by 100, respectively. | |||||||||||||||||||||||||||||||||||
| Instability Index: | |||||||||||||||||||||||||||||||||||
| The instability index, an estimate of peptide stability, is calculated as follows: | |||||||||||||||||||||||||||||||||||
where L is the length of peptide and DIWV from the study by Guruprasad et al. is an instability weight value of a dipeptide starting at position i . Peptides with II values greater than 40 are considered to be unstable. | |||||||||||||||||||||||||||||||||||
| Hydropathicity: | |||||||||||||||||||||||||||||||||||
| Grand average of hydropathicity index (GRAVY) is used to represent the hydrophobicity value of a peptide, which calculates the sum of the hydropathy values of all the amino acids divided by the sequence length. GRAVY was calculated using the hydropathy values from Kyte and Doolittle. | |||||||||||||||||||||||||||||||||||
| Search | |||||||||||||||||||||||||||||||||||
ADAM offers multiple search capabilities, which can be classified into two basic categories: sequence search and structural search. One can search what kind of information an AMP sequence belongs to (sequence search) or which related sequences are in a unique AMP structure or structural fold cluster (structural search). | |||||||||||||||||||||||||||||||||||
Sequence search: | |||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||
Structural search: | |||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||
| Browse clusters | |||||||||||||||||||||||||||||||||||
To examine AMP sequence-structure relationships, ADAM allows users to browse through the fold clusters built using TM-score. Each fold cluster gethers together the AMP structures with the same structral fold defined by TM-score. Each cluster would list the number of AMP sequences and the Pfam sequence associated with the fold. A representative structure would be chosen by PDBselect to display its CATH and SCOP annotation (if any). Any structures related to a particular fold defined by CATH or SCOP can be displayed through their hyperlinks. More detail information for the AMP sequences in the cluster can be displayed by clicking the cluster icon. Here each structure in the cluster would be annotated by CATH, SCOP, and Pfam (if any). | |||||||||||||||||||||||||||||||||||
| Prediction | |||||||||||||||||||||||||||||||||||
In the ADAM database, we provide two different tools to examine whether the peptides or proteins are antimicrobial. Users can upload protein sequences with FASTA format to ADAM to make prediction. | |||||||||||||||||||||||||||||||||||
SVM: | |||||||||||||||||||||||||||||||||||
Support vector machines are used to analyze AMP data and recognize patterns. Users are allowed to paste sequences in FASTA format to run our SVM models. | |||||||||||||||||||||||||||||||||||
HMM: | |||||||||||||||||||||||||||||||||||
HMMER is used to search any AMP homologs in the given protein sequence. It implements a probabilistic model called profile hidden Markov models (profile HMMs) to make predictions. Users are allowed to enter sequences in FASTA format to run our HMM models. | |||||||||||||||||||||||||||||||||||
| Links | |||||||||||||||||||||||||||||||||||
| Other database links: | |||||||||||||||||||||||||||||||||||